Our Methods
Spatial omics
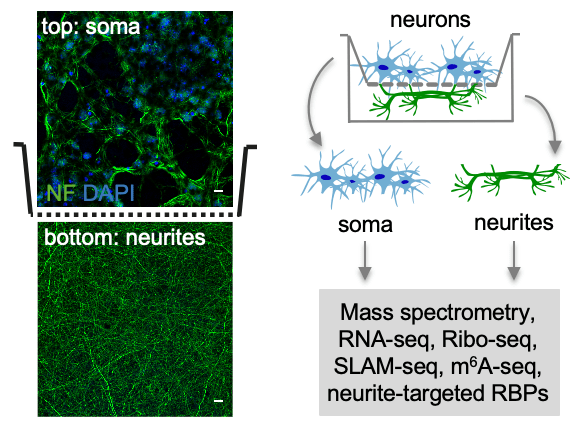
To study RNA localization in neurons, we developed an approach that allows us to separate neurons on subcellular compartments – soma and neurites – for omics analyses (spatial omics, Zappulo et al., Nature Comm 2017; Ludwik et al. Methods 2019). For that, neurons are plated on a microporous membrane so that soma stays on top, and neurites grow through the pores on the lower side of the membrane. Soma and neurites are then collected from the different sides of the membrane and used for RNA-seq, mass-spectrometry, Ribo-seq, or other omics analyses. This approach can be applied to different types of neurons, from stem cell-derived (Zappulo et al. Nature Comm 2017) to primary cortical neurons isolated from the mouse brain (Ciolli Mattioli et al. NAR 2019). It is easily scalable and produces homogeneous material. In contrast, material captured during laser dissection is heterogenous, containing numerous non-neuronal cell types.
Ribosome profiling
Ribosome profiling, or Ribo-seq, is a method that generates ribosome footprints on mRNA and thereby allows to study translation. mRNA in cell lysate is digested by RNAse I, and fragments protected by ribosomes from the digestion – ribosome footprints – are isolated and sequenced. For spatial ribosome profiling, we optimized the protocol for low amounts of material obtainable from neurites (Zappulo et al. Nature Comm 2017). In particular, we showed that ribosome-footprinted fragments could be isolated by electrophoresis-based size selection, and the ribosome purification step can be skipped without compromising the quality of analysis.
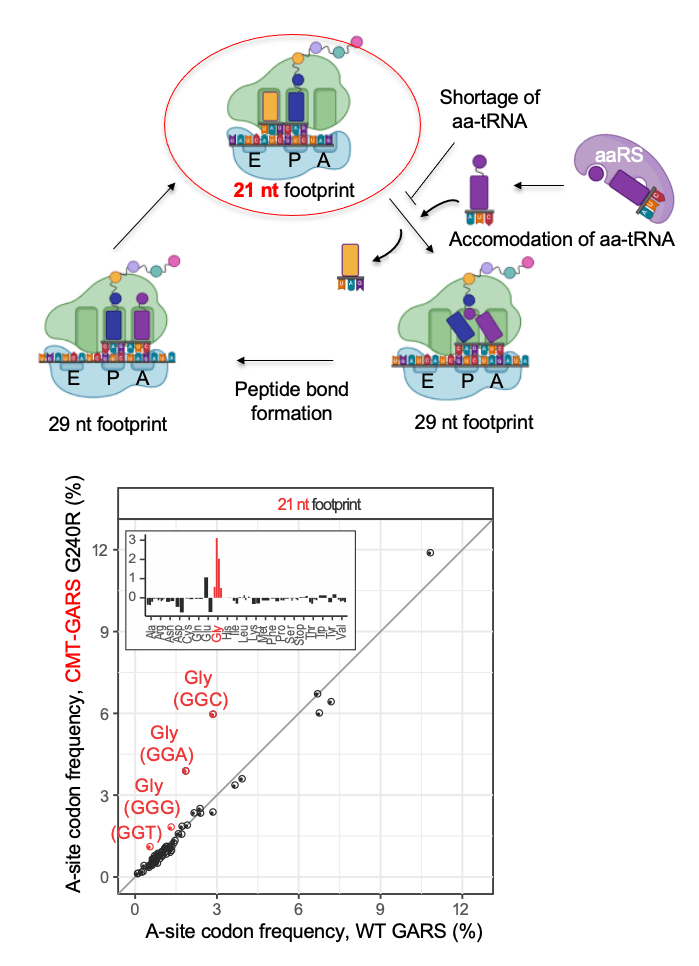
We also used high-resolution ribosome profiling to distinguish between different functional states of the ribosome – before and after binding of amino acyl-tRNA into the A-site of the ribosome. Due to conformational changes of the ribosome during elongation, ribosomes lacking tRNA in their A-sites generate shorter 21-22 nt ribosome footprints than ribosomes with occupied A-sites (27-29 nt footprints). Therefore, high–resolution ribosome profiling can detect ribosome pausing on mRNA due to the lack of a specific amino acyl-tRNA. For example, we used this approach to show that mutation in glycyl-tRNA synthase, causative of Charcot–Marie–Tooth disease, leads to ribosome pausing on glycine codons (Mendonsa et al. NAR 2021).
Imaging
smFISH
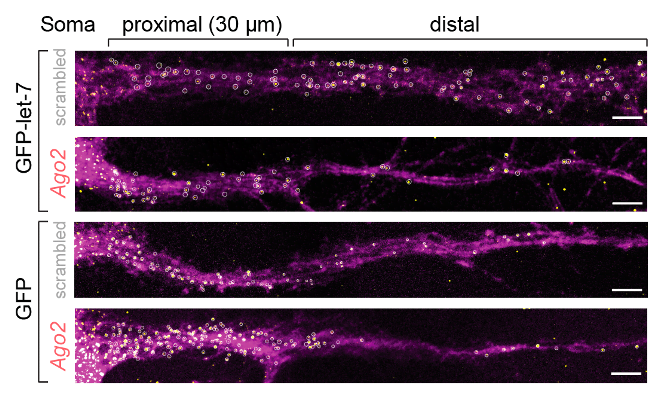
To monitor mRNA localization and local translation in cells, we also use imaging. For RNA visualization, we rely on smFISH (single molecule fluorescent in situ hybridization). For example, we used this method to show that splicing isoforms of the small GTPase Cdc42 are localized to distinct neuronal compartments (Ciolli Mattioli et al. NAR 2019) and to demonstrate the role of let-7 binding site in mRNA localization in primary cortical neurons (Mendonsa et al. Nature Neuroscience 2023).
puro-PLA
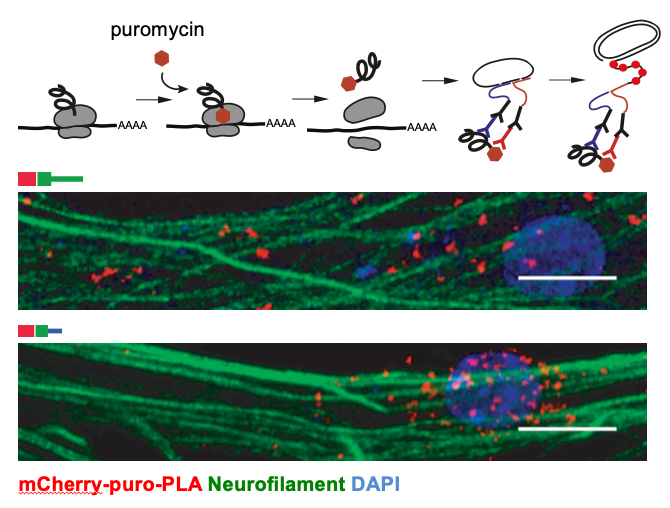
To assess local translation, we apply puro-PLA (tom Dieck et al. 2015). This method allows the visualization of newly synthesized proteins via puromycin-tagging. Puromycin is a structural analog of the aminoacylated 3′end of tRNA, which is incorporated into the nascent polypeptide chains, resulting in puromycin fusion proteins. In the puro-PLA assay, puromycin-tagging is combined with the proximity-ligation assay (PLA) to detect the spatial coincidence of two antibodies: (1) an anti-puromycin antibody and (2) an antibody against a specific protein of interest. For example, we used this approach to show translation of selected neurite-localized mRNAs (Zappulo et al. Nature Comm 2017) and to demonstrate the role of Cdc42 alternative 3’UTRs in local translation (Ciolli Mattioli et al. NAR 2019).
Test systems
To study mechanisms of RNA localization, we use two main test systems: mouse primary cortical neuron culture and neurons derived from hiPSC lines.
Primary cortical neuron
We prepare primary cortical neuron cultures according to the accepted procedures (Kaech and Banker, 2006) and co-culture neurons with astrocytes. We use primary cortical neurons for separation on soma and neurites (Ciolli Mattioli et al., NAR 2019; Mendonsa et al., Nature Neuroscience 2023), imaging and other experiments. An essential advantage of primary neurons over neuronal cell lines and stem cell-derived neurons is that they allow us to study RNA localization mechanisms in vivo. For example, we identified let-7 binding site as de novo zipcode in primary cortical neurons (Mendonsa et al., Nature Neuroscience 2023), but not in neuroblastoma cell lines CAD and N2A. This is due to differences in miRNA expression profiles between primary neurons and neuronal cell lines.
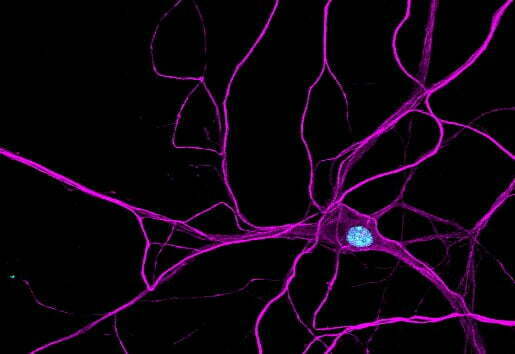
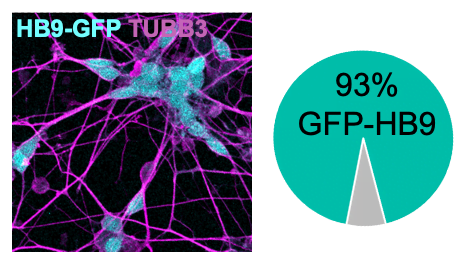
Stem cell-derived neurons
To study neurodegenerative diseases, such as ALS, we use neurons derived from patient hiPSCs (human induced pluripotent stem cells). To generate neurons from hiPSCs, we introduce a cassette for the inducible expression of neurogenic transcription factors in their genome. As ALS affects motor neurons (i.e., neurons that produce motion), we express transcription factors specific to motor neurons (Hester et al. 2011; Son et al. 2011 and others). Within three weeks of induction, these hiPSCs differentiate into neurons that express synaptic and motor neuron markers (e.g., CHAT, Synapsin, and others). Unlike traditional protocols relying on small molecules to induce differentiation, this approach results in a highly homogenous population of neurons (> 90 % motor neurons). An advantage of hiPSC-derived motor neurons over mouse primary motor neurons is that they allow us to obtain neurons with the patient genetic background.
Other methods
We also apply other transcriptomic methods in our analysis. For example, we performed SLAM-seq and small RNA-seq to analyze the role of mRNA stability (Herzog et al., 2017) and miRNAs in subcellular localization of mRNAs.
For genetic editing of hiPSC lines for ALS modeling, we rely on CRISPR-Cas. For example, we generated isogenic controls for patient-derived hiPSC lines, where disease-causing mutations are corrected to the wild-type. Such isogenic lines are essential negative controls for ALS modeling experiments.